



Among the five gene therapies currently on the market in the US, the FDA has required immunogenicity testing, whether a laboratory developed test (LDT) or a companion diagnostic (CDx), as a condition to approval for the three most recently approved: etranacogene dezaparvovec (Hemgenix®), delandistrogene moxeparvovec-rokl (Elevidys®), and valoctocogene roxparvovec-rvox (Roctavian®). Given the agency’s emphasis on testing for pre-existing immunity, gene therapy companies are increasingly partnering with diagnostic developers to develop immune assays.

The path from immunogenicity assay development to CDx approval for adeno-associated virus (AAV) gene therapy is complex and time-consuming because so much of the assay validation must be done sequentially. To ensure that the right assay is ready at the right time at every stage of a clinical program, it is essential for gene therapy developers to plan ahead.
In this blog, we discuss 8 best practices for AAV gene therapy assay development and CDx co-development.
1. Choose the most appropriate assay
There are two primary methods for detecting pre-existing humoral immunogenicity to gene therapy vectors: total binding antibody (TAb) assays and neutralizing antibody (NAb) assays (see Table 2). Key considerations when selecting an immunogenicity assay include:
- Assay throughput. TAb assays are one-day assays that can accommodate many samples per plate. NAb assays are typically multi-day assays that require more replicates and controls, thus allowing fewer samples per plate.
- Sensitivity. TAb assays are generally more sensitive, though assay development, validation, and platform all impact sensitivity. Assay sensitivity will determine seroprevalence so, in any given population, differing assays, even those of the same type, can give markedly different seroprevalence results depending on underlying sensitivity.
- Selectivity. As a cell-based assay, NAb assays will be more prone to interference by endogenous factors such as hemoglobin, lipids, and certain medications than TAb assays.
- Reagent requirements. TAb assays often utilize an empty capsid or the drug substance, while NAb assays require a reporter vector that involves a separate manufacturing process.
- Correlation between assays. Only a subset of binding antibodies exert neutralization. Thus, a TAb assay could potentially pick up antibodies that may not have a clinical effect. On the other hand, non-antibody factors may inhibit transduction so a NAb assay may pick up artifacts that do not have clinical relevance.
- Format. Both TAb and NAb assays can be designed to be either qualitative or semi-quantitative. With a qualitative assay, the result is either a positive or negative response based on signal relative to background. If used for screening, a qualitative assay would exclude any patient with an antibody response above the limit of detection, making the fewest patients eligible for therapy. With a semi-quantitative assay, the result is a titer value, which allows for correlation between a specific antibody titer and the efficacy of the therapeutic. Semi-quantitative values also enable gene therapy developers to set a data-driven cutoff for enrollment.

2. Carefully select the clinical cutoff
Unlike an analytical cut point for anti-drug antibody (ADA) assays, a clinical cutoff for an immunogenicity assay is a result which drives a medical decision. For a qualitative assay, the cutoff is essentially the limit of detection. For a semi-quantitative assay, the cutoff should be a specific titer greater than the minimum required dilution. Further, the assay validation to support clinical trial use will involve a panel of samples including two that are within 20% of the clinical cutoff, one above and one below. For a clinical diagnostic, these samples must be human in origin. Generating these samples can be a significant challenge, particularly for cell-based NAb assays that often have ~20% variability. Thus, it is essential to have a defined release process when preparing these samples.
3. Plan ahead for critical reagent requirements
Given the expectation that the assay will be used for many years, it is critical for gene therapy and diagnostic developers to ensure a reliable source of critical reagents such as reporter vectors, cell lines, negative matrices, and positive controls.
- For an early-phase clinical trial assay (CTA), a single reagent lot should be sufficient and good manufacturing practice (GMP) is not required.
- For a pivotal study CTA, multiple lots are generally required and, while GMP is not a mandate, having GMP material at this stage would be advantageous.
- For an in vitro diagnostic (IVD) study to support a premarket approval (PMA) submission, at least three reagent lots will be needed. If the material is not GMP, then it must be as close to GMP as possible with rigorous documentation and testing. Precision recommends manufacture in a facility compliant with a good quality management system (QMS) and the material should be accompanied by a certificate of analysis.
4. Develop a global regulatory strategy for clinical trial assay (CTA) testing and associated regulatory timelines
Start with a risk assessment of the CTA internally based on how the assay will be used as part of the trial, for example, if the assay is being used for inclusion/exclusion. Risk will impact both regulatory requirements and the level of assay validation required. Figure 1 provides an overview of the US regulatory requirements -based on the results of the risk assessment.

Questions to think through when assessing risk include:
- Is the assay being used prospectively or retrospectively?
- How is the assay being used? For research, stratification, or inclusion/exclusion?
- Has the assay ever been used in prior investigations with safety data?
- What type of sample is required, and does it involve an invasive procedure?
- If a fresh biopsy is required, is it a significant risk procedure?
- Are the test results used to determine whether the patient receives treatment? If so:
- Would a false-positive result lead to a patient not receiving a known and effective therapy or standard of care (SOC)?
- How does the safety profile of the experimental therapeutic compare to SOC?
- Have patients exhausted all SOC options?
In the United States (US), the early-stage risk assessment methodologies available include assessment by an institutional review board (IRB), which serves as a surrogate for the FDA, submission of a study risk determination (SRD) Q-Submission to the FDA, or assumption of high risk and submission of an IDE application. When selecting a risk assessment methodology, it is important to consider timing for submission development and FDA review periods. For example, for the SRD Q-Submission process should take 4- 6 weeks to develop, and FDA has up to 90 days to review the document and provide the assessment. The Pre-IDE Q-Submission and pre-IDE meeting with FDA allows review and meeting timeline of 75 days for FDA. Further, building realistic timelines that allows for each step of the process for submission develop and regulatory Agency review is critical.
Keep in mind that assay validation requirements differ between the US and EU. Under the In Vitro Diagnostic Regulation (IVDR), if an assay has a medical purpose and its result impacts the medical management of a patient, the develop must comply with IVDR Annex XIV and Article 58(2). Challenges associated with gene therapy CTAs in the EU include unclear validation requirements, lack of a pre-submission process, differences in requirements among member states, and long timelines for Annex XIV submission review and approval, all of which should be factored into a global regulatory strategy. Therefore, the internal timelines should have parallel workstreams for regulatory submissions in the different global regions.
5. Allow time for the IDE enabling analytical validation studies
Clinical Laboratory Improvement Amendments (CLIA) requirements may not be robust enough to support an IDE application. Often, studies included in an IDE application for a gene therapy CTA require more samples or replicates, more rigorous assessment of endogenous and exogenous interference, and evaluation of sample and control stability. The Pre-IDE Q-Submission process will allow collaboration with the Agency prior to conducting the testing to agree upon the level of analytical validation to support the IDE application. These studies typically align more closely to the CLSI guidelines. The analytical validation package for the final PMA will require a robust data package that aligns more closely with the recommendations in the CLSI guidelines, and these studies will take time to complete. Planning ahead is key for conducting the studies and potentially repurposing some of the data from the IDE application into the final analytical data package for the PMA with agreement from FDA during the pre-submission process.
6. Avoid screening in early phase studies if possible
Using the CTA for inclusion/exclusion in Phase 1 studies dramatically affects the timing for assay development and validation as well as the associated regulatory submissions which can delay the start of the trial as these will need to be completed before the Phase 1 study (see Figure 2). Without efficacy data to support selection of a clinical cutoff, it may be required to set the most conservative cutoff (the limit of detection), and the associated CLIA validation to support an IDE application will need to be conducted prior to the Phase 1 study. Further, the Dx Sponsor may need to participate in the Pre-IDE Q-submission process that can take up to 5 months to complete from submission development through meeting with FDA. Then the CLIA validation studies must be conducted by the Dx Sponsor prior to drafting the IDE and approval of the IDE application to enable testing in the trial can take 30 FDA review days. While Q-Submissions in general are not required by the FDA, they provide opportunities for developers to communicate and collaborate with the Agency to glean intelligence for their assay development programs, especially for an IDE application where the requirements for the analytical validation data package are not published.
If possible, using an all comers testing strategy would be preferred in Phase 1 to allow selection of a cutoff based on clinical efficacy. Further, this will allow the necessary time for the regulatory submissions and communications to take place prior to the Phase 2.
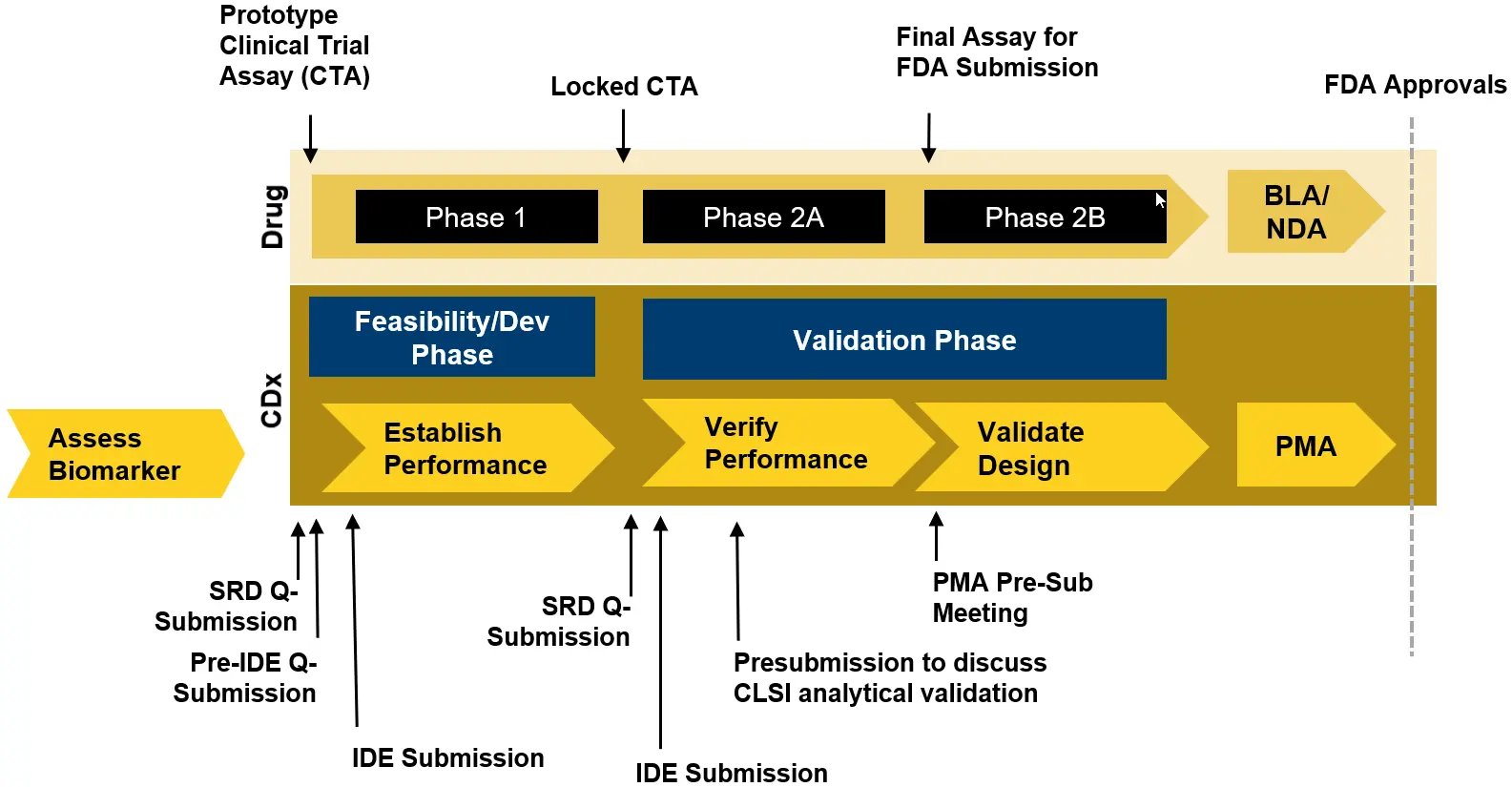
7. Avoid bridging studies
Using a final locked CTA in the Phase 3 registrational trial maximizes the likelihood of drug/CDX co-approval and eliminates the need for bridging studies, which can cause significant delays.
8. Select a diagnostic partner with the right scientific, regulatory, and quality credentials
Choosing a companion diagnostic partner early and wisely is key. Ideally, the diagnostic partner has the capability to support development from assay selection through commercialization since switching labs midstream will require repeat assay development and performance validation. To succeed in a new, rapidly developing field involving complex assays, that partner must have deep laboratory experience in assay optimization and CLSI validation and extensive regulatory and quality expertise. Ensure they have the right instrumentation, software, and infrastructure to support the studies in all geographies.
At Precision for Medicine, we provide global support for complex innovations through seven specialty labs in North American and Europe, six sample processing labs, and over 3000 employees. Our IVD and CDx team has supported over 100 CDx regulatory filings in countries around the world. We have supported more than 15 AAV-focused gene therapy companies and their projects across AAV serotypes, assay types, and therapeutic areas, including rare diseases. Precision for Medicine also developed, validated, and serves as the sole site for the LDT for Hemgenix®.


